




De origem congênita, a cranioestenose é uma doença que causa o fechamento precoce das linhas de sutura, que unificam as placas ósseas do crânio. Ela é diagnosticada ainda nos primeiros meses de vida de um bebê, e conta com tratamento cirúrgico seguro.

O que é?
Ao contrário do que algumas pessoas pensam, o crânio não é um único osso, pois ele é formado por placas ósseas. Esses ossos se juntam por meio de linhas de sutura. As linhas de sutura, por sua vez, facilitam o desenvolvimento cerebral de uma criança durante a fase de crescimento. Contudo, é possível que aja uma junção prematura desses ossos, o que pode causar desde problemas no desenvolvimento do cérebro até alterações de formato do crânio, o que é conhecido como cranioestenose ou craniossinostose.
Existem cinco tipos de placas ósseas no crânio, são elas:
- Osso frontal: localizado na parte da frente da cabeça.
- Ossos parietais: localizados nas laterais do crânio.
- Occipital: localizado na parte de trás da cabeça.
- Osso temporal: um par de ossos que acomodam o aparelho auditivo.
Sendo assim, existem diferentes tipos de craniossinostoses, dependendo da sutura cranial afetada. São elas:
- Escafocefalia: também chamada de clinocefalia, esse tipo de cranioestenose é o mais comum. É caracterizada pelo crânio exageradamente alongado no sentido ântero posterior.
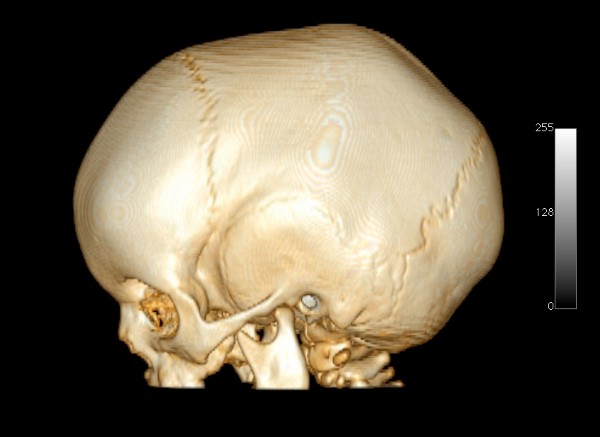
- Plagiocefalia Anterior: corresponde ao fechamento precoce da sutura coronal unilateral. É caracterizada por uma assimetria do crânio e da face, além de ter um crescimento anormal do lado acometido, com deslocamento do nariz e um abaulamento frontal contralateral à sutura comprometida.
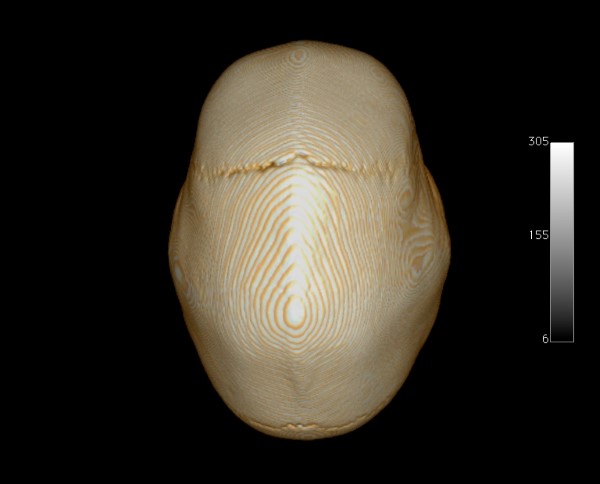
- Trigonocefalia: ela corresponde ao fechamento precoce da sutura metópica. A principal característica é a fronte estreita, marcada por uma crista mediana e apontada anteriormente como a proa de um barco, levando a uma forma triangular. É a cranioestenose de sutura única mais associada a anomalias cromossômicas.
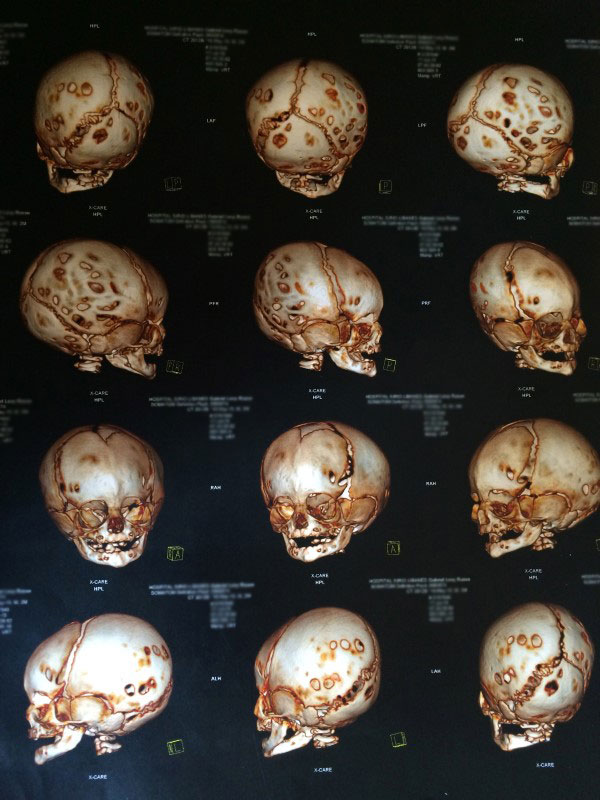
- Braquicefalia é o fechamento precoce das duas suturas coronais. A característica mais comum é o crânio retraído anterior e posteriormente, com a parte de trás levemente alongada.
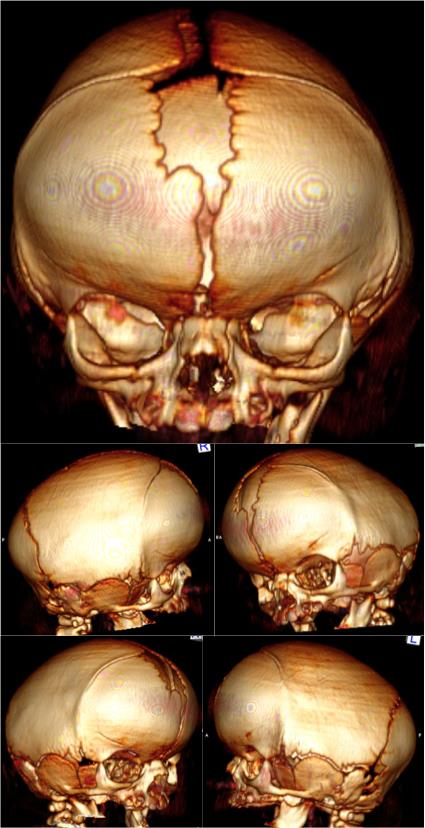
A doença tem origem congênita, ou seja, é adquirida antes do nascimento. A principal ameaça para às crianças portadoras de cranioestenose é algo chamado de hipertensão intracraniana, que pode causar desde um pequeno descompasso mental até um déficit cognitivo ou mesmo perda da visão.
Quais as causas?
A doença é classificada em duas categorias: sindrômica e não sindrômica. A primeira é relacionada às síndromes, como é o caso das que comprometem tecidos como músculos e ossos e até mesmo órgãos, como o coração.
A segunda tem origem ligada apenas à malformação craniana. Apesar de algumas vezes as causas serem idiopáticas (desconhecidas ou não aparentes) é possível citar três fatores que podem facilitar o surgimento da craniossinostose no período de desenvolvimento do embrião:
- Hereditariedade: no caso de síndrome, há a influência dos genes de cada família. Dessa forma, se o pai ou outro parente teve a cranioestenose, a criança também pode desenvolvê-la.
- Intrauterino: o ambiente onde o feto se desenvolve é comprimido, e de acordo com a pressão presente, pode haver mudança no formato do crânio.
Quem faz parte do grupo de risco?
Pesquisas indicam que a incidência da anomalia é de 1 em cada 2000 crianças, mas que em uma contagem de 10000 nascimentos, esse número pode girar em torno de 14 bebês. Em relação aos sexos, meninos tem mais chance de desenvolver a doença que meninas. A relação, nesse caso, é de dois para um. Apesar de ainda estar na fase de estudos, alguns autores explicam que devido à restrição de espaço no útero, pode haver aumento da incidência em gêmeos.
O ideal é que o diagnóstico seja feito no nascimento da criança. Apesar disso, a natureza do parto pode dificultar a identificação da doença. Dessa forma, é importante ficar atento nos dois meses posteriores ao nascimento, pois alguns casos necessitam de tratamento cirúrgico, que deve ser realizado até o sexto mês de vida.
Como é feito o diagnóstico?
O diagnóstico é feito parcialmente por um pediatra, que logo após o nascimento pode confirmar ou não a suspeita de craniossinostose. Para tanto, ele pode realizar o exame clínico no intuito de encontrar suturas que fecharam precocemente. Elas podem ser identificadas facilmente, pois causam um crescimento descompensado das placas ósseas.
Se a suspeita for confirmada, ele pode encaminhar o pequeno paciente para um neurocirurgião, a fim de uma avaliação mais especializada. Se o diagnóstico for feito em um curto período, a chance de haver sequelas na criança diminui drasticamente, e ela poderá então viver com qualidade de vida e sem necessidades especiais.
Para confirmar a suspeita de cranioestenose, é importante realizar alguns exames que permitam, além de constatar o fechamento precoce das linhas de sutura, encontrar possíveis alterações nos ossos faciais. Os exames mais usados são:
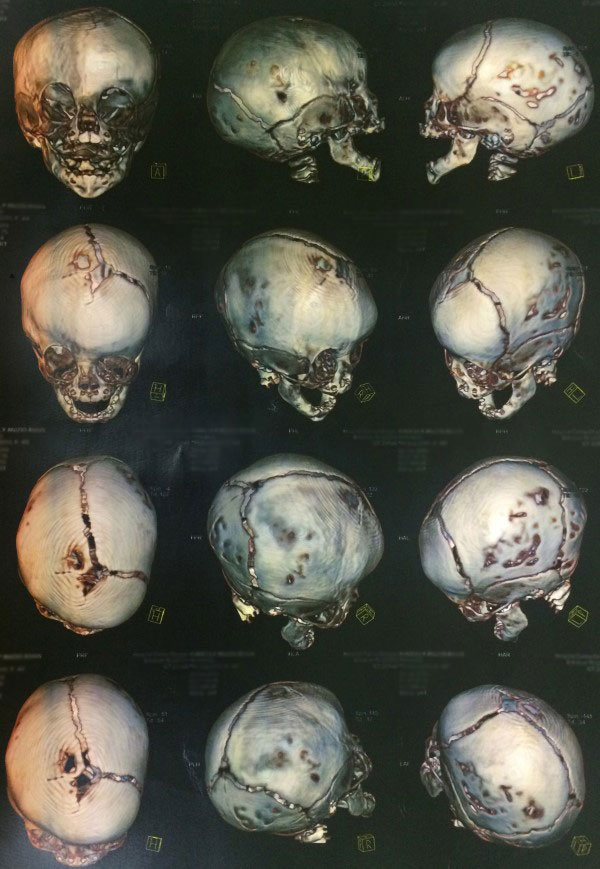
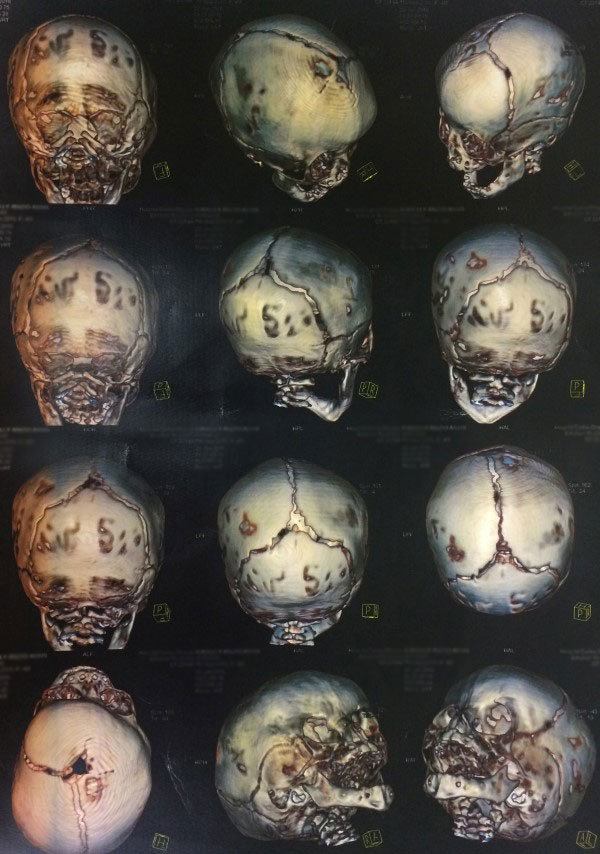
- Tomografia computadorizada de crânio com reconstrução óssea tridimensional: permite visualizar o crânio como um todo e assim, identificar possíveis falhas e suturas fechadas antes da hora. A tomografia permite uma excelente análise das modificações da base do crânio e das órbitas, sobretudo nos raros casos em que o exame físico apresenta alterações ou ainda quando há suspeita de fechamento de mais de uma sutura.
- Ressonância Magnética Encefálica: maior sensibilidade para detectar alterações no crânio e no encéfalo, como as suturas precoces. O exame se utiliza de campos magnéticos e ondas de rádio para dar forma e criar imagens computadorizadas com grande definição.
- Radiografia: apesar de ser menos pedido, o exame auxilia a visualizar as estruturas ósseas. Normalmente ele é utilizado na ortopedia para verificar traumas ou fraturas ósseas, mas no caso da craniossinostose, visa exclusivamente verificar anormalidades nas placas ósseas.
Quais são as formas de tratamento?
Se o diagnóstico for identificado como sendo o de cranioestenose, o tratamento adequado para a anomalia é exclusivamente a cirurgia. A primeira intervenção cirúrgica de uma cranioestenose que se tem conhecimento foi feita em 1890 por Marie-Lannelongue. Desde então, várias técnicas foram desenvolvidas, diminuindo significativamente o impacto sobre os bebês portadores da anomalia.
Para o procedimento, o neurologista, juntamente ao pediatra, deverão orientar os pais e familiares presentes sobre todas as etapas da cirurgia de forma simples e acolhedora.
A cirurgia visa eliminar as suturas que se fecharam precocemente e então remodelar o crânio, de modo que o aspecto deformado suma. Vale lembrar que ela não vai criar uma nova sutura. No caso de a sutura se encontrar fechada, um novo centro de crescimento ósseo não pode ser criado. Em suma, a operação restabelece a forma e o tamanho adequado do crânio.
Alguns centros especializados acreditam que não há necessidade de operar caso haja apenas uma sutura fechada precocemente. Entretanto, é importante lembrar que a área de anomalia do crânio fará com que ocorra um aumento na pressão intracraniana e inclusive prejudicar o fluxo sanguíneo.
Além do aspecto de saúde do paciente, há também o aspecto social, pois mesmo que o impacto nas funções cerebrais seja mínimo, as deformidades cranianas podem ter um efeito profundo sobre aparência e personalidade. O convívio social e inclusive o ato de ir à escola podem ser profundamente afetados. Por isso é importante realizar a cirurgia mesmo que seja apenas uma sutura acometida.
Informações sobre pós-operatório e prevenção
A cirurgia deve ser feita em um grande centro hospitalar com experiência na área e que conte com uma UTI (Unidade de Terapia Intensiva) pediátrica. Além disso, a equipe médica tem de ser preparada e contar com um anestesista responsável e habituado a trabalhar com crianças. A duração do procedimento cirúrgico gira em torno de duas a seis horas, dependendo das necessidades de remodelagem da caixa craniana.
No mais, o pós-operatório é tranquilo e sem complicações severas. O risco de haver sequela por conta da cirurgia é muito baixo, pois o procedimento requer apenas a remodelagem do crânio, descartando a possibilidade de acessar o cérebro.
O ano posterior à cirurgia requer acompanhamento médico constante. Após esse período, os retornos são anuais, até que a partir dos sete anos a criança tem alta.









